About Me

Hi there! I'm Claire, a researcher based in the Department of Statistics at the University of Oxford. I am a member of the Oxford Protein Informatics Group (OPIG), supervised by Prof. Charlotte Deane.
I am the group's lead Research Software Engineer - my main job is maintaining our suite of antibody tools, known the SAbDab-SAbPred platform. I am responsible for facilitating in-house installation of this platform for any pharmaceutical companies wishing to use them, and also look after our web servers.
Research
As Research Software Engineer for the Oxford Protein Informatics Group (OPIG), most of my time these days is spent maintaining the group's software. In particular I look after the tools related to our antibody research, known as the SAbDab-SAbPred platform. Many pharmaceutical companies use these tools, and I facilitate this by installing the platform in-house through consultancy. The tools are also available online - I developed the websites for SAbDab,, SAbPred, and most recently OAS.
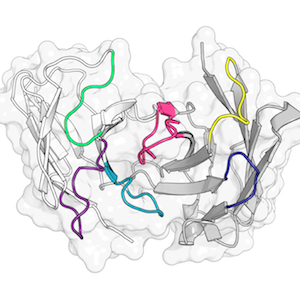
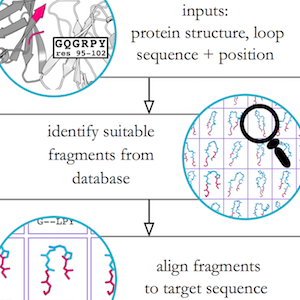
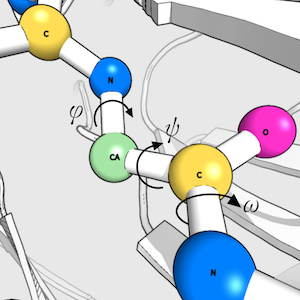
When I do have the chance to do my own research, my work focusses on protein structure prediction, - mainly of loops, which are the regions of a protein that are not part of secondary structure elements. Loops often play a key role in protein function; understanding their properties and knowing their structures is therefore extremely valuable.

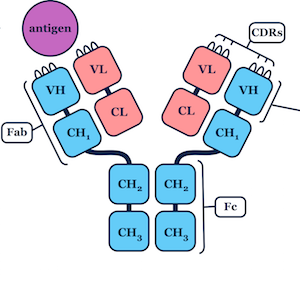
I developed a novel algorithm for the prediction of protein loop structures, called Sphinx. Sphinx uses a combination of knowledge-based and ab initio approaches to maximise our use of the available structural data. While it was originally designed to improve our ability to model the CDR H3 loops of antibodies, I have also developed versions for membrane protein loops and loops from non-specific protein types. I have also explored ways to improve the prediction of long loops, through the inclusion of properties that can be predicted from sequence. For example, I have used contact prediction to establish sets of spatial constraints, which can then be considered during decoy ranking to improve predictions.
More recently, I have carried out analyses of loops which are able to adopt multiple conformations, and investigated the ability of several algorithms to predict their structures. Unfortunately, it seems that current methodologies are accurate for loops of a single conformation, but fail when they attempt to model conformational ensembles. For more information on this, or my other research, see my Publications list!
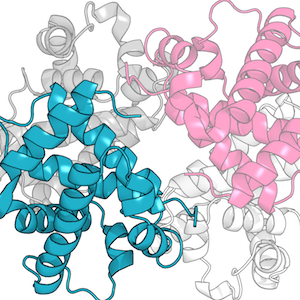
Through student supervision I also work/have worked on the prediction of therapeutic antibody developability, structural analysis of immune repertoires, antibody humanisation, improving loop decoy ranking through deep learning, and predicting antibody binding.
Publications
Background
Research Software Engineer
University of Oxford, 2018-Present
Responsibilities: Maintaining the group's software, including web servers and our virtual machine SAbBox; consultancy work for pharmaceutical companies (e.g. installation of/assistance using our tools, research projects); student supervision; my own research sometimes!
Postdoctoral Researcher and Associate Director of the Systems Approaches to Biomedical Science CDT
University of Oxford, 2016-2018
Main research themes: analysis and structure prediction of protein loops with multiple conformations; membrane protein loop modelling; analysis of therapeutic antibodies (through supervision).
DPhil Systems Approaches to Biomedical Science
University of Oxford, 2012-2016
Thesis title: Hybrid methods for protein loop modelling
MChem (Master of Chemistry), First Class
Durham University, 2007-2011
Masters project title: Simulating photodissociation reactions of protonated aminophenols
Dissertation title: Cellular drug delivery
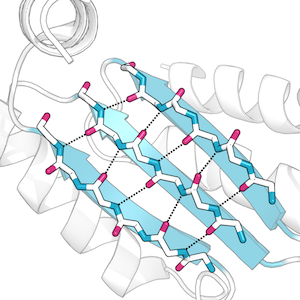
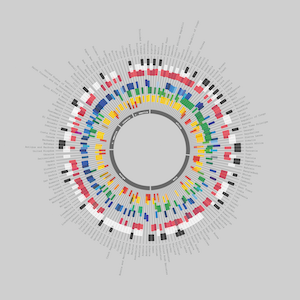
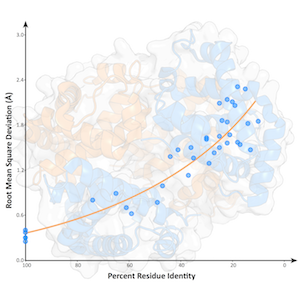
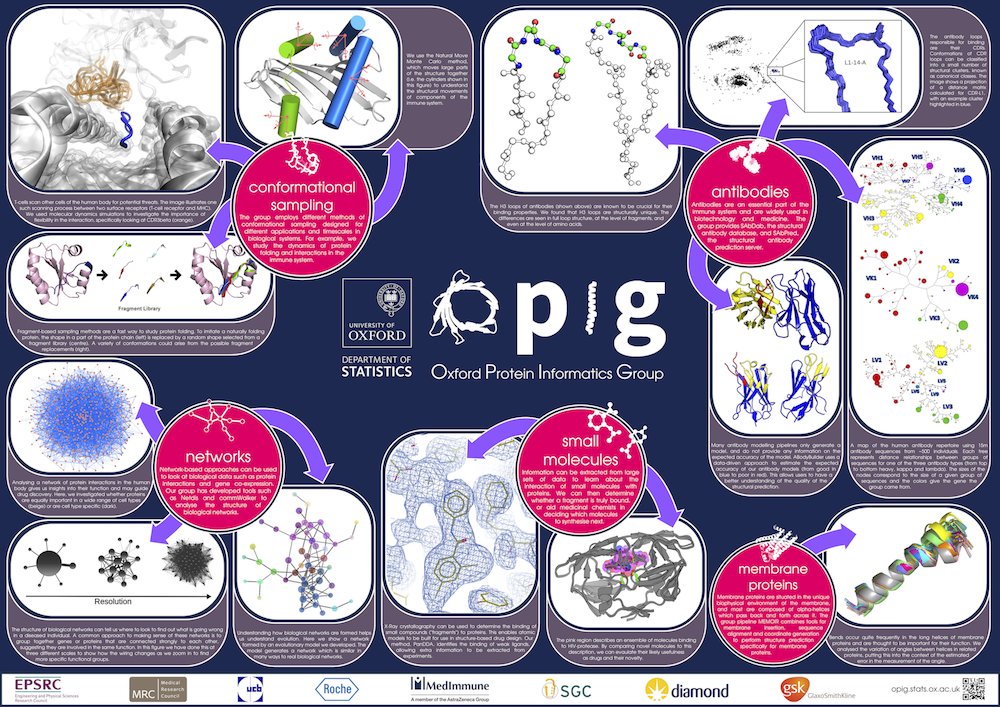
Gallery
PyMOL images
Selected paper/thesis figures
Other
Posters
Other
Besides science, I enjoy:
- Music - I play clarinet, saxophone and piano and am a member of the OUP Orchestra
- Art and Crafts - particularly pencil drawing and crocheting
- Reading
- Cryptic Crosswords
- Baking
- Swimming

Contact
- Email me at claire.marks{at}stats.ox.ac.uk
- Find me on LinkedIn